Benchmark: delnx vs R glmGamPoi¶
glmGamPoi is a fantastic package, but unfortunately it is in R. delnx reimplements glmGamPoi’s quasi-likelihood NB GLM approach in python with JAX. This notebook validates that the two implementations agree on real data, and benchmarks runtime on simulated data.
import time
import delnx as dx
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
from scipy.stats import pearsonr
1. Concordance on real data¶
We compare delnx and R glmGamPoi on a GLI3 KO pseudobulk dataset (28 samples, 16k genes) to verify that dispersions, coefficients, and p-values agree.
adata = sc.read_h5ad("data/GLI3_KO_45d_pseudobulk.h5ad")
adata.obs["GLI3_KO"] = adata.obs["GLI3_KO"].astype(str)
counts = np.round(np.asarray(adata.layers["counts"])).astype(int)
adata.X = counts.astype(np.float64)
print(adata)
print(f"Conditions: {adata.obs['GLI3_KO'].value_counts().to_dict()}")
AnnData object with n_obs × n_vars = 28 × 16199
obs: 'psbulk_replicate', 'cell_type', 'organoid', 'GLI3_KO', 'psbulk_cells', 'psbulk_counts', 'size_factor'
var: 'dispersion', 'dispersion_deseq', 'dispersion_mle', 'dispersion_edger', 'mean', 'mean_norm'
uns: 'log1p'
layers: 'counts', 'psbulk_props'
Conditions: {'True': 16, 'False': 12}
fit_dx = dx.tl.nb_fit(adata, condition_key="GLI3_KO", reference="True")
res_dx = dx.tl.nb_test(adata, fit_dx, contrast="GLI3_KO[T.False]")
print(f"Significant genes (padj < 0.05): {(res_dx['padj'] < 0.05).sum()}")
res_dx.head()
INFO Fitting 16199 genes with 2 coefficient(s) (batch_size=512)
INFO Applying quasi-likelihood shrinkage
Significant genes (padj < 0.05): 181
| feature | log2fc | coef | stat | pval | padj | |
|---|---|---|---|---|---|---|
| 0 | EMX1 | 6.761568 | 4.686762 | 83.710401 | 4.326881e-10 | 0.000007 |
| 1 | ZNF429 | -5.335782 | -3.698482 | 75.925150 | 1.246159e-09 | 0.000010 |
| 2 | SFTA3 | -8.920224 | -6.183028 | 60.244111 | 1.368526e-08 | 0.000074 |
| 3 | CXXC4 | -0.873021 | -0.605132 | 54.949587 | 3.389955e-08 | 0.000106 |
| 4 | ZIC5 | 1.904016 | 1.319763 | 54.774321 | 3.496747e-08 | 0.000106 |
import rpy2.robjects as ro
from rpy2.robjects import numpy2ri, pandas2ri, default_converter
from rpy2.robjects.packages import importr
converter = default_converter + numpy2ri.converter + pandas2ri.converter
_ctx = converter.context()
_ctx.__enter__()
base = importr("base")
glmGamPoi = importr("glmGamPoi")
print(f"R version: {base.R_version_string[0]}")
R version: R version 4.5.3 (2026-03-11)
ro.globalenv["counts"] = counts.T
ro.globalenv["condition"] = ro.FactorVector(
adata.obs["GLI3_KO"].values, levels=ro.StrVector(["True", "False"]))
ro.globalenv["gene_names"] = ro.StrVector(adata.var_names.tolist())
ro.globalenv["sample_names"] = ro.StrVector(adata.obs_names.tolist())
ro.r('''
rownames(counts) <- gene_names
colnames(counts) <- sample_names
col_data <- data.frame(GLI3_KO = condition, row.names = sample_names)
fit_gp <- glm_gp(counts, design = ~ GLI3_KO, col_data = col_data,
size_factors = "normed_sum", overdispersion_shrinkage = TRUE)
res_gp <- test_de(fit_gp, contrast = cond(GLI3_KO = "False") - cond(GLI3_KO = "True"))
''')
res_glmgp = pd.DataFrame({
"feature": np.array(ro.r("res_gp$name")),
"log2fc": np.array(ro.r("res_gp$lfc")),
"pval": np.array(ro.r("res_gp$pval")),
"padj": np.array(ro.r("res_gp$adj_pval")),
})
disp_glmgp = np.array(ro.r("fit_gp$overdispersions"))
print(f"Significant genes (padj < 0.05): {(res_glmgp['padj'] < 0.05).sum()}")
res_glmgp.head()
Significant genes (padj < 0.05): 187
| feature | log2fc | pval | padj | |
|---|---|---|---|---|
| 0 | LRP6 | -0.293760 | 0.007480 | 0.204735 |
| 1 | PDCL2 | -0.445917 | 0.447408 | 0.855558 |
| 2 | PRSS56 | -0.820560 | 0.189395 | 0.723788 |
| 3 | ZNF665 | 0.306711 | 0.138165 | 0.670246 |
| 4 | WDR3 | -0.083624 | 0.433192 | 0.847047 |
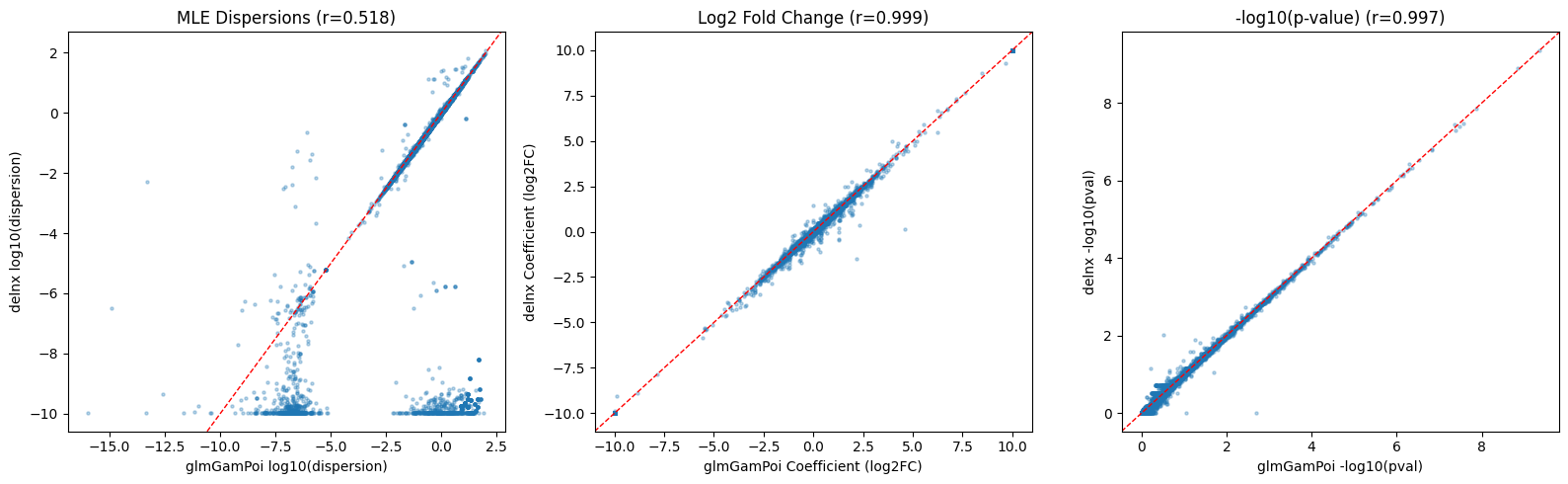
fig, axes = plt.subplots(1, 3, figsize=(16, 5))
# Dispersions
ax = axes[0]
d1, d2 = fit_dx.overdispersions, disp_glmgp
mask = (d1 > 0) & (d2 > 0) & np.isfinite(d1) & np.isfinite(d2)
ax.scatter(np.log10(d2[mask]), np.log10(d1[mask]), s=5, alpha=0.3)
ax.axline((0, 0), slope=1, color="red", linestyle="--", linewidth=1)
r, _ = pearsonr(np.log10(d2[mask]), np.log10(d1[mask]))
ax.set_xlabel("glmGamPoi log10(dispersion)")
ax.set_ylabel("delnx log10(dispersion)")
ax.set_title(f"MLE Dispersions (r={r:.3f})")
# LFC
ax = axes[1]
lfc_dx = res_dx.set_index("feature")["log2fc"]
lfc_gp = res_glmgp.set_index("feature")["log2fc"]
common = lfc_dx.index.intersection(lfc_gp.index)
l1, l2 = lfc_dx[common].dropna(), lfc_gp[common].dropna()
common2 = l1.index.intersection(l2.index)
r, _ = pearsonr(l1[common2], l2[common2])
ax.scatter(l2[common2], l1[common2], s=5, alpha=0.3)
ax.axline((0, 0), slope=1, color="red", linestyle="--", linewidth=1)
ax.set_xlabel("glmGamPoi Coefficient (log2FC)")
ax.set_ylabel("delnx Coefficient (log2FC)")
ax.set_title(f"Log2 Fold Change (r={r:.3f})")
# P-values
ax = axes[2]
p_dx = res_dx.set_index("feature")["pval"]
p_gp = res_glmgp.set_index("feature")["pval"]
p1, p2 = p_dx[common].dropna(), p_gp[common].dropna()
common3 = p1.index.intersection(p2.index)
p1v, p2v = p1[common3].replace(0, np.nan).dropna(), p2[common3].replace(0, np.nan).dropna()
common4 = p1v.index.intersection(p2v.index)
r, _ = pearsonr(-np.log10(p1v[common4]), -np.log10(p2v[common4]))
ax.scatter(-np.log10(p2v[common4]), -np.log10(p1v[common4]), s=5, alpha=0.3)
ax.axline((0, 0), slope=1, color="red", linestyle="--", linewidth=1)
ax.set_xlabel("glmGamPoi -log10(pval)")
ax.set_ylabel("delnx -log10(pval)")
ax.set_title(f"-log10(p-value) (r={r:.3f})")
plt.tight_layout()
plt.show()
plt.close()
padj_dx = res_dx.set_index("feature")["padj"]
padj_gp = res_glmgp.set_index("feature")["padj"]
common_padj = padj_dx.dropna().index.intersection(padj_gp.dropna().index)
sig_dx = set(common_padj[padj_dx[common_padj] < 0.05])
sig_gp = set(common_padj[padj_gp[common_padj] < 0.05])
jaccard = len(sig_dx & sig_gp) / len(sig_dx | sig_gp) if len(sig_dx | sig_gp) > 0 else 0
print(f"delnx significant: {len(sig_dx)}")
print(f"glmGamPoi significant: {len(sig_gp)}")
print(f"Shared: {len(sig_dx & sig_gp)}")
print(f"Jaccard index: {jaccard:.3f}")
delnx significant: 181
glmGamPoi significant: 187
Shared: 181
Jaccard index: 0.968
delnx and glmGamPoi show very similar results, with high agreement in p-values and coefficients, even though the dispersion estimates vary for some genes.
2. Runtime benchmarks¶
We benchmark delnx vs R glmGamPoiruntime across two axis: the number of cells/samples per condition, and the number of conditions. We also include a “cold start” line for delnx, which includes the JIT compilation time.
import anndata as ad
def simulate_pseudobulk(n_genes, n_conditions, n_samples_per_condition, mean_counts=100, dispersion=0.5, seed=42):
"""Simulate pseudobulk NB counts for a multi-condition experiment."""
rng = np.random.default_rng(seed)
n_samples = n_conditions * n_samples_per_condition
gene_means = rng.lognormal(np.log(mean_counts), 1.0, size=n_genes)
size_factors = rng.lognormal(0, 0.2, size=n_samples)
# Vectorized NB sampling
r = 1.0 / dispersion
mu = gene_means[None, :] * size_factors[:, None] # (n_samples, n_genes)
p = r / (r + mu)
counts = rng.negative_binomial(r, p)
# Condition labels
labels = [f"cond_{i}" for i in range(n_conditions) for _ in range(n_samples_per_condition)]
adata_sim = ad.AnnData(X=counts.astype(np.float64))
adata_sim.var_names = [f"gene_{i}" for i in range(n_genes)]
adata_sim.obs_names = [f"sample_{i}" for i in range(n_samples)]
adata_sim.obs["condition"] = labels
return adata_sim
print("Simulation function defined")
Simulation function defined
# Measure cold start (JIT compilation) time
t0 = time.time()
adata_warmup = simulate_pseudobulk(1000, 2, 3)
fit_warmup = dx.tl.nb_fit(adata_warmup, condition_key="condition", reference="cond_0", verbose=False)
_ = dx.tl.nb_test(adata_warmup, fit_warmup, contrast="condition[T.cond_1]")
jit_warmup_time = time.time() - t0
print(f"JIT warmup (cold start): {jit_warmup_time:.1f}s")
JIT warmup (cold start): 7.5s
2.1 Sample scaling (pseudobulk → single-cell)¶
This covers the typical two-group DE test (e.g., treatment vs control) as sample size grows. Lower sample numbers cover (pseudo)bulk scenarios, while scaling to higher sample numbers enbales testing on the single-cell level.
N_GENES = 10000
samples_per_cond = [5, 20, 100, 1000, 5000, 10000, 30000]
sample_results = []
for n_spc in samples_per_cond:
n_total = 2 * n_spc
print(f"\n--- {n_spc:,} samples/condition, {n_total:,} total ---")
adata_sim = simulate_pseudobulk(N_GENES, 2, n_spc)
# delnx
t0 = time.time()
fit_sim = dx.tl.nb_fit(adata_sim, condition_key="condition", reference="cond_0", verbose=False)
_ = dx.tl.nb_test(adata_sim, fit_sim, contrast="condition[T.cond_1]")
t_dx = time.time() - t0
sample_results.append({"samples_per_cond": n_spc, "total_samples": n_total,
"method": "delnx", "time": t_dx})
print(f" delnx: {t_dx:.1f}s")
# R glmGamPoi
c_sim = np.round(np.asarray(adata_sim.X)).astype(int)
ro.globalenv["sim_counts"] = c_sim.T
ro.globalenv["sim_cond"] = ro.FactorVector(
adata_sim.obs["condition"].values, levels=ro.StrVector(["cond_0", "cond_1"]))
ro.globalenv["sim_genes"] = ro.StrVector(adata_sim.var_names.tolist())
ro.globalenv["sim_samples"] = ro.StrVector(adata_sim.obs_names.tolist())
t0 = time.time()
ro.r('''
rownames(sim_counts) <- sim_genes; colnames(sim_counts) <- sim_samples
sim_col <- data.frame(condition = sim_cond, row.names = sim_samples)
sim_fit <- glm_gp(sim_counts, design = ~ condition, col_data = sim_col,
size_factors = "normed_sum", overdispersion_shrinkage = TRUE)
sim_res <- test_de(sim_fit, contrast = cond(condition = "cond_1") - cond(condition = "cond_0"))
''')
t_gp = time.time() - t0
sample_results.append({"samples_per_cond": n_spc, "total_samples": n_total,
"method": "glmGamPoi", "time": t_gp})
print(f" glmGamPoi: {t_gp:.1f}s")
sample_df = pd.DataFrame(sample_results)
pivot = sample_df.pivot_table(index="samples_per_cond", columns="method", values="time")
print("\n" + pivot.to_string())
--- 5 samples/condition, 10 total ---
delnx: 0.5s
glmGamPoi: 5.1s
--- 20 samples/condition, 40 total ---
delnx: 3.2s
glmGamPoi: 7.1s
--- 100 samples/condition, 200 total ---
delnx: 8.5s
glmGamPoi: 9.6s
--- 1,000 samples/condition, 2,000 total ---
delnx: 13.7s
glmGamPoi: 32.2s
--- 5,000 samples/condition, 10,000 total ---
delnx: 30.6s
glmGamPoi: 108.2s
--- 10,000 samples/condition, 20,000 total ---
delnx: 33.0s
glmGamPoi: 200.7s
--- 30,000 samples/condition, 60,000 total ---
delnx: 64.3s
glmGamPoi: 574.1s
method delnx glmGamPoi
samples_per_cond
5 0.480144 5.105261
20 3.242199 7.134030
100 8.483462 9.596197
1000 13.667955 32.178085
5000 30.623963 108.160478
10000 33.006560 200.650563
30000 64.259846 574.115171
fig, ax = plt.subplots(figsize=(8, 5))
for method, style in [("delnx", dict(color="#1f77b4", marker="o")), ("glmGamPoi", dict(color="#2ca02c", marker="^"))]:
sub = sample_df[sample_df["method"] == method].sort_values("samples_per_cond")
ax.plot(sub["samples_per_cond"], sub["time"] / 60, label=method, linewidth=2, markersize=8, **style)
# Also plot cold start as separate line
sub = sample_df[sample_df["method"] == "delnx"].sort_values("samples_per_cond")
ax.plot(sub["samples_per_cond"], (sub["time"] + jit_warmup_time) / 60, color="#1f77b4", linestyle="--", linewidth=1, alpha=0.5,
label=f"delnx cold start (approximately + {jit_warmup_time:.0f}s)")
ax.set_xlabel("Samples per condition")
ax.set_ylabel("Time (minutes)")
ax.set_title(f"Single DE test: sample scaling ({N_GENES:,} genes, 2 conditions)")
# Scale x axis logarithmically
ax.set_xscale("log")
ax.legend()
ax.grid(True, alpha=0.3)
plt.tight_layout()
plt.show()
plt.close()
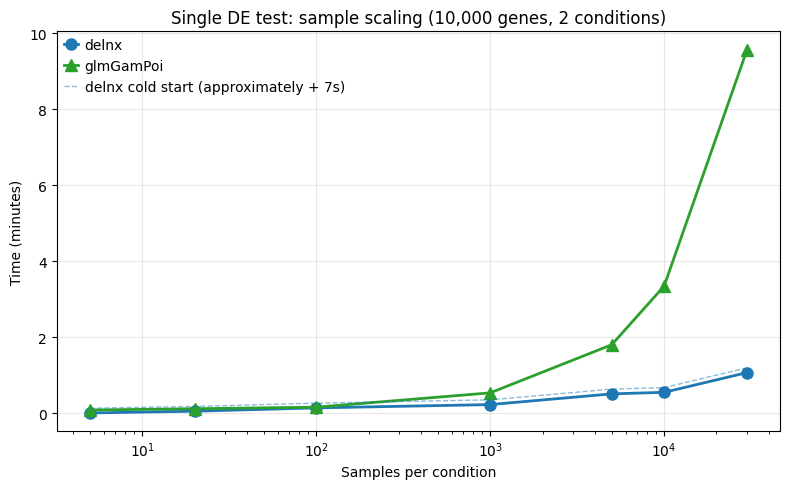
This is where delnx scales very well, thanks to JAX’s efficient vectorization.
2.2 Condition scaling¶
Here we test scaling over the number of conditions, which is relevant for multi-condition DE and perturbation screens. We simulate a dataset with 10k genes and 100 samples, and vary the number of conditions from 4 to 100.
N_GENES = 10000
condition_counts = [4, 10, 25, 50, 100]
screen_results = []
for n_cond in condition_counts:
N_REPS = 1000 // n_cond # keep total samples ~1000 for faster benchmarking
n_total = n_cond * N_REPS
n_coef = n_cond # intercept + (n_cond - 1) treatment contrasts = n_cond
print(f"\n--- {n_cond} conditions, {n_total} samples, {n_coef} coefficients ---")
adata_sim = simulate_pseudobulk(N_GENES, n_cond, N_REPS)
# delnx: fit once, test all contrasts
t0 = time.time()
fit_sim = dx.tl.nb_fit(adata_sim, condition_key="condition", reference="cond_0", verbose=False)
contrasts = [c for c in fit_sim.design_column_names if c != "Intercept"]
for c in contrasts:
_ = dx.tl.nb_test(adata_sim, fit_sim, contrast=c)
t_dx = time.time() - t0
screen_results.append({"n_conditions": n_cond, "n_samples": n_total, "n_coef": n_coef,
"method": "delnx", "time": t_dx})
print(f" delnx: {t_dx:.1f}s (fit + {len(contrasts)} contrasts)")
# R glmGamPoi: fit once, test all contrasts
c_sim = np.round(np.asarray(adata_sim.X)).astype(int)
ro.globalenv["scr_counts"] = c_sim.T
cond_levels = [f"cond_{i}" for i in range(n_cond)]
ro.globalenv["scr_cond"] = ro.FactorVector(
adata_sim.obs["condition"].values, levels=ro.StrVector(cond_levels))
ro.globalenv["scr_genes"] = ro.StrVector(adata_sim.var_names.tolist())
ro.globalenv["scr_samples"] = ro.StrVector(adata_sim.obs_names.tolist())
ro.globalenv["scr_n_cond"] = n_cond
t0 = time.time()
ro.r('''
rownames(scr_counts) <- scr_genes; colnames(scr_counts) <- scr_samples
scr_col <- data.frame(condition = scr_cond, row.names = scr_samples)
scr_fit <- glm_gp(scr_counts, design = ~ condition, col_data = scr_col,
size_factors = "normed_sum", overdispersion_shrinkage = TRUE)
for (i in 1:(scr_n_cond - 1)) {
cname <- paste0("cond_", i)
scr_res <- test_de(scr_fit, contrast = cond(condition = cname) - cond(condition = "cond_0"))
}
''')
t_gp = time.time() - t0
screen_results.append({"n_conditions": n_cond, "n_samples": n_total, "n_coef": n_coef,
"method": "glmGamPoi", "time": t_gp})
print(f" glmGamPoi: {t_gp:.1f}s")
screen_df = pd.DataFrame(screen_results)
pivot = screen_df.pivot_table(index="n_conditions", columns="method", values="time")
print("\n" + pivot.to_string())
--- 4 conditions, 1000 samples, 4 coefficients ---
delnx: 1.2s (fit + 3 contrasts)
glmGamPoi: 26.8s
--- 10 conditions, 1000 samples, 10 coefficients ---
delnx: 2.2s (fit + 9 contrasts)
glmGamPoi: 39.5s
--- 25 conditions, 1000 samples, 25 coefficients ---
delnx: 6.6s (fit + 24 contrasts)
glmGamPoi: 72.0s
--- 50 conditions, 1000 samples, 50 coefficients ---
delnx: 31.8s (fit + 49 contrasts)
glmGamPoi: 139.2s
--- 100 conditions, 1000 samples, 100 coefficients ---
delnx: 224.1s (fit + 99 contrasts)
glmGamPoi: 320.6s
method delnx glmGamPoi
n_conditions
4 1.234963 26.774637
10 2.248468 39.521930
25 6.589387 72.025133
50 31.801404 139.168610
100 224.106427 320.590345
fig, ax = plt.subplots(figsize=(8, 5))
for method, style in [("delnx", dict(color="#1f77b4", marker="o")), ("glmGamPoi", dict(color="#2ca02c", marker="^"))]:
sub = screen_df[screen_df["method"] == method].sort_values("n_conditions")
ax.plot(sub["n_conditions"], sub["time"] / 60, label=method, linewidth=2, markersize=8, **style)
sub = screen_df[screen_df["method"] == "delnx"].sort_values("n_conditions")
ax.plot(sub["n_conditions"], (sub["time"] + jit_warmup_time) / 60, color="#1f77b4", linestyle="--", linewidth=1, alpha=0.5, label=f"delnx cold start (+{jit_warmup_time:.0f}s)")
ax.set_xlabel("Number of conditions")
ax.set_ylabel("Total time (minutes)")
ax.set_title(f"Perturbation screen: joint model ({N_GENES:,} genes, {N_REPS} replicates/condition)")
ax.legend()
ax.grid(True, alpha=0.3)
plt.tight_layout()
plt.show()
plt.close()
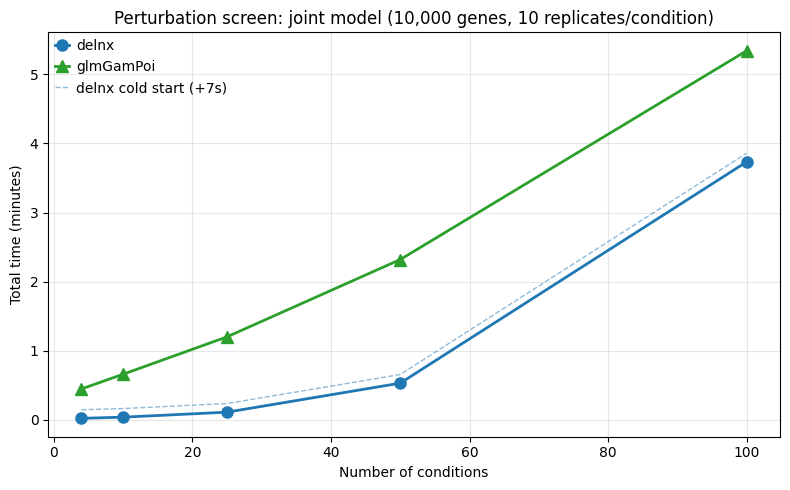
Here, delnx is much faster for small numbers of conditions, but the gap stays roughly constant as conditions increase.