Negative binomial DE with quasi-likelihood dispersion shrinkage¶
delnx implements GPU-accelerated negative binomial GLMs following the glmGamPoi approach with quasi-likelihood dispersion shrinkage for differential expression analysis. The two-step workflow (nb_fit + nb_test) separates model fitting from hypothesis testing, allowing you to reuse a single fit across multiple contrasts.
import delnx as dx
import scanpy as sc
import numpy as np
import matplotlib.pyplot as plt
# Load example pseudobulk data
adata = sc.read_h5ad("data/GLI3_KO_45d_pseudobulk.h5ad")
adata.obs["GLI3_KO"] = adata.obs["GLI3_KO"].astype(str) # Ensure string type for design matrix
# Use raw counts (X contains normalized values)
adata.X = np.round(np.asarray(adata.layers["counts"])).astype(np.float64)
print(adata)
print(f"Count range: {adata.X.min():.0f} - {adata.X.max():.0f}")
AnnData object with n_obs × n_vars = 28 × 16199
obs: 'psbulk_replicate', 'cell_type', 'organoid', 'GLI3_KO', 'psbulk_cells', 'psbulk_counts', 'size_factor'
var: 'dispersion', 'dispersion_deseq', 'dispersion_mle', 'dispersion_edger', 'mean', 'mean_norm'
uns: 'log1p'
layers: 'counts', 'psbulk_props'
Count range: 0 - 123900
Fit the model¶
nb_fit handles everything in one call: size factor estimation, dispersion MLE with Cox-Reid bias adjustment, quasi-likelihood shrinkage, and coefficient fitting via IRLS.
# Fit negative binomial GLMs with quasi-likelihood dispersion shrinkage
fit = dx.tl.nb_fit(adata, condition_key="GLI3_KO", reference="True")
print(f"Coefficients: {fit.design_column_names}")
print(f"Genes fitted: {len(fit.overdispersions)}")
print(f"Dispersion range: {fit.overdispersions.min():.4f} - {fit.overdispersions.max():.2f}")
INFO Fitting 16199 genes with 2 coefficient(s)
INFO Applying quasi-likelihood shrinkage
Coefficients: ['Intercept', 'GLI3_KO[T.False]']
Genes fitted: 16199
Dispersion range: nan - nan
fig, axes = plt.subplots(1, 2, figsize=(14, 5))
# Left: dispersion distribution
valid_disp = fit.overdispersions[fit.overdispersions > 0]
axes[0].hist(np.log10(valid_disp), bins=50, color="steelblue", alpha=0.7, edgecolor="white")
axes[0].set_xlabel("log10(dispersion)")
axes[0].set_ylabel("Number of genes")
axes[0].set_title("Dispersion distribution")
axes[0].axvline(np.log10(np.median(valid_disp)), color="red", linestyle="--", label=f"median={np.median(valid_disp):.3f}")
axes[0].legend()
# Right: mean-dispersion relationship
gene_means = np.asarray(adata.X).mean(axis=0) if hasattr(adata.X, 'toarray') else adata.X.mean(axis=0)
mask = (gene_means > 0) & (fit.overdispersions > 0)
axes[1].scatter(np.log10(gene_means[mask]), np.log10(fit.overdispersions[mask]), s=5, alpha=0.3)
axes[1].set_xlabel("log10(mean expression)")
axes[1].set_ylabel("log10(dispersion)")
axes[1].set_title("Mean-dispersion relationship")
plt.tight_layout()
plt.show()
plt.close()
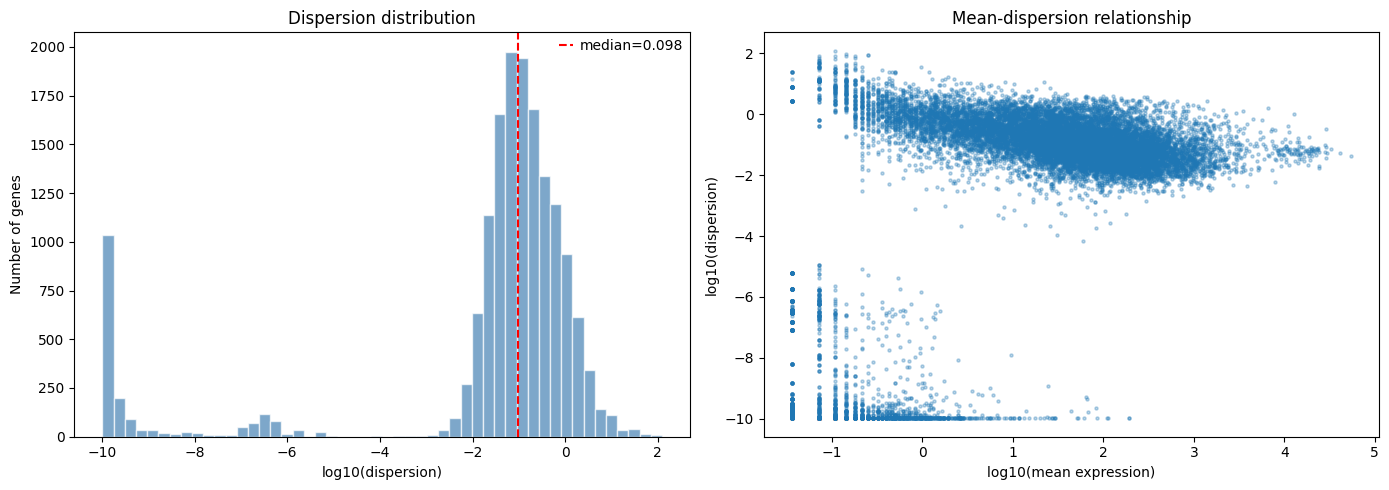
The left panel shows the distribution of MLE dispersions across genes. Most genes cluster around the median, with a long tail of highly variable genes.
The right panel shows the characteristic mean-dispersion relationship: lowly expressed genes tend to have higher dispersion, while highly expressed genes converge toward lower dispersion values.
Test for differential expression¶
Now we test for DE using the fitted model. nb_test uses a quasi-likelihood F-test, which accounts for the uncertainty in dispersion estimation.
# Test for differential expression
de_results = dx.tl.nb_test(adata, fit, contrast="GLI3_KO[T.False]")
print(de_results)
feature log2fc coef stat pval padj
0 EMX1 6.761568 4.686762 83.710401 4.326881e-10 0.000007
1 ZNF429 -5.335782 -3.698482 75.925150 1.246159e-09 0.000010
2 SFTA3 -8.920224 -6.183028 60.244111 1.368526e-08 0.000074
3 CXXC4 -0.873021 -0.605132 54.949587 3.389955e-08 0.000106
4 ZIC5 1.904016 1.319763 54.774321 3.496747e-08 0.000106
... ... ... ... ... ... ...
16194 GJB3 NaN NaN NaN NaN 1.000000
16195 CCL16 NaN NaN NaN NaN 1.000000
16196 CCR1 NaN NaN NaN NaN 1.000000
16197 PLET1 NaN NaN NaN NaN 1.000000
16198 ORM2 NaN NaN NaN NaN 1.000000
[16199 rows x 6 columns]
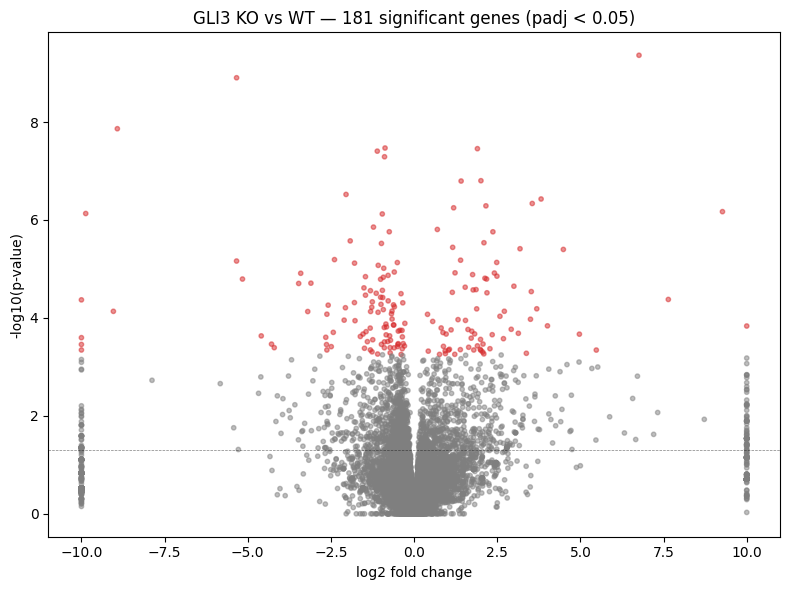
# Volcano plot
fig, ax = plt.subplots(figsize=(8, 6))
sig = de_results["padj"] < 0.05
colors = np.where(sig, "tab:red", "tab:grey")
ax.scatter(de_results["log2fc"], -np.log10(de_results["pval"]), c=colors, s=10, alpha=0.5)
ax.axhline(-np.log10(0.05), color="black", linestyle="--", linewidth=0.5, alpha=0.5)
ax.set_xlabel("log2 fold change")
ax.set_ylabel("-log10(p-value)")
ax.set_title(f"GLI3 KO vs WT — {sig.sum()} significant genes (padj < 0.05)")
plt.tight_layout()
plt.show()
plt.close()